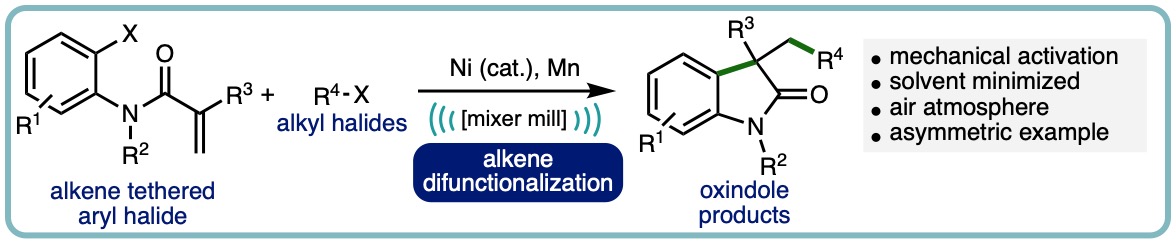
Nickel-Catalyzed Intramolecular Alkene Difunctionalization by Ball-Milling
A mechanochemical nickel-catalyzed intramolecular difunctionalization reaction of alkene tethered aryl halides with alkyl halides is herein described. This method allows for synthesis of 3,3-disubstituted heterocycles, namely oxindoles, with shorter reaction times than solution-phase counterparts. Additionally, this process is solvent minimized, with DMA used in liquid-assisted grinding (LAG) quantities and circumvents the need for chemical activation of the terminal reductant (manganese) through mechanical grinding. The process can be scaled up to yield over a gram of product and modest enantioinduction is possible by utilizing a chiral PyrOx ligand. (Adv. Synth. Catal., 2023, 365, 1477-1484) [link] [Hot Topic: Earth-Abundant Transition Metal Catalysis]
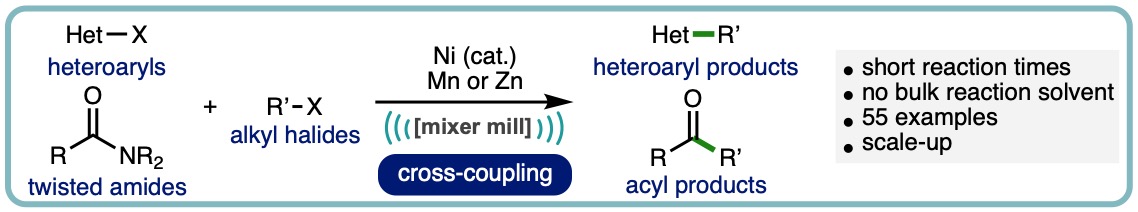
Mechanical Activation of Zero-Valent Metal Reductants for Nickel-Catalyzed Cross-Electrophile Coupling
The cross-electrophile coupling of either twisted-amides or heteroaryl halides with alkyl halides, enabled by ball-milling, is herein described. The operationally simple nickel-catalysed process has no requirement for inert atmosphere or dry solvents and delivers the corresponding acylated or heteroarylated products across a broad range of substrates. Key to negating the necessity of inert reaction conditions is the mechanical activation of the raw metal terminal reductant; manganese in the case of twisted amides and zinc for heteroaryl halides. (ACS Catal., 2022, 12, 13681-13689. [link]
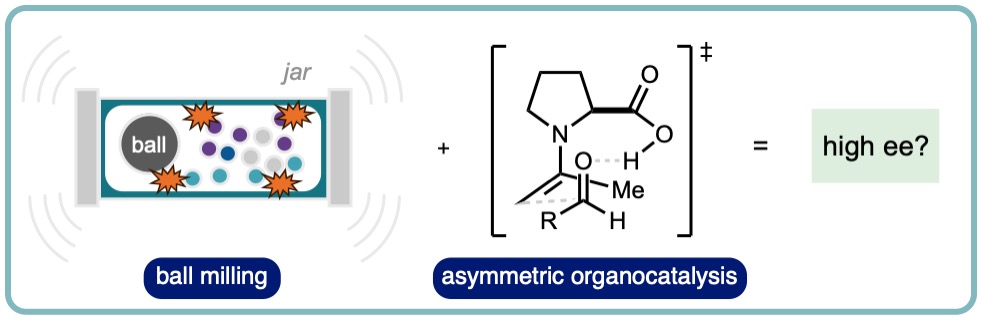
Mechanochemical Organocatalysis: Do High Enantioselectivities Contradict what we Might Expect?
Ball mills input energy to samples by pulverizing the contents of the jar. Each impact on the sample or wall of the jar results in an instantaneous transmission of energy in the form of a temperature and pressure increase (volume reduction). Conversely, enantioselective organocatalytic reactions proceed through perceived delicate and well organized transition states. Does there exist a dichotomy in the idea of enantioselective mechanochemical organocatalysis? This review provides a survey of the literature reporting the combination of organocatalytic reactions with mechanochemical ball milling conditions. Where possible, direct comparisons of stirred in solution, stirred neat and ball milled processes are drawn with a particular focus on control of stereoselectivity. (ChemSusChem, 2022, 15, e202102157 [link])
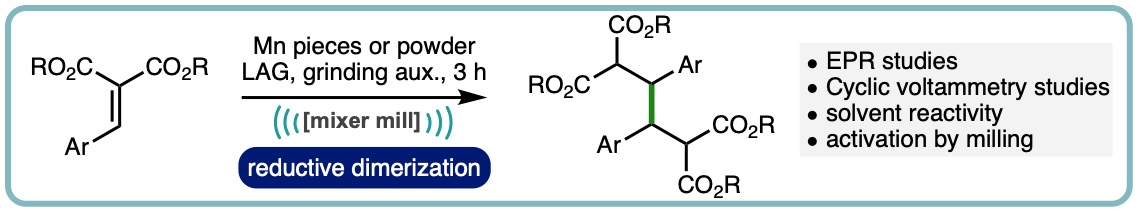
Ball Milling Enabled Reactivity of Manganese Metal
Efforts to generate organomanganese reagents under ball milling conditions have led to the serendipitous discovery that manganese metal can mediate the reductive dimerization of arylidene malonates. The newly uncovered process has been optimized and its mechanism explored using CV measurements, radical trapping experiments, EPR spectroscopy and solution control reactions. This unique reactivity can also be translated to solution where upon premilling of the manganese is required. (Angew. Chem. Int. Ed., 2021, 60, 23128-23133. [link])
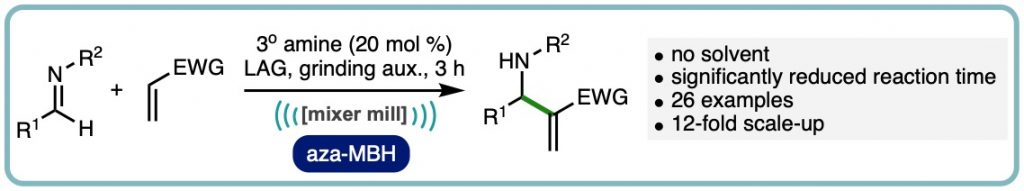
Expedient Organocatalytic Aza-Morita-Baylis-Hillman Reaction Through Ball-Milling
A ball-milling enabled tertiary amine catalysed aza-Morita-Baylis-Hillman reaction is reported. The reaction process does not require solvent, has significantly shorter reaction times than previous methods, and is reported on a range of imines and acrylate Michael acceptors across more than 25 examples. A 12-fold scaled-up example is also reported as well as experimental comparisons to solution based experiments and neat-stirred reactions including (ACS Sustainable Chem. Eng., 2020, 8, 17876-17881. [link])
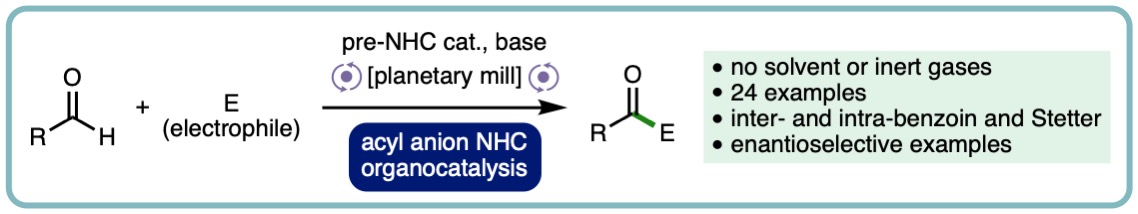
N-Heterocyclic Carbene Acyl Anion Organocatalysis by Ball-Milling
The ability to conduct N-Heterocyclic carbene catalysed acyl anion chemistry under ball-milling conditions is reported for the first time. This process has been exemplified through applications to intermolecular-benzoin, intramolecular-benzoin, intermolecular-Stetter and intramolecular-Stetter reactions including asymmetric examples and demonstrates that this mode of mechanistically complex organocatalytic reaction can operate under solvent minimised conditions (ChemSusChem, 2019, 13, 131-135. [link] [Hot Topic: Organocatalysis]).