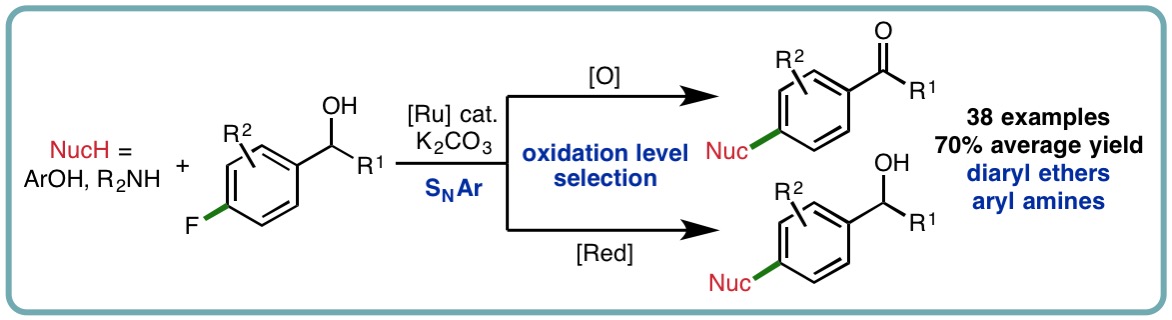
Borrowing Hydrogen for Organic Synthesis
Borrowing hydrogen is a process that is used to diversify the synthetic utility of commodity alcohols. A catalyst firstly oxidizes an alcohol by removing hydrogen to form a reactive carbonyl compound. This intermediate can undergo a diverse range of subsequent transformations before the catalyst returns the “borrowed” hydrogen to liberate the product and regenerate the catalyst. In this way, alcohols may be used as alkylating agents whereby the sole byproduct of this one-pot reaction is water. In recent decades, significant advances have been made in this area, demonstrating many effective methods to access valuable products. This outlook highlights the diversity of metal and biocatalysts that are available for this approach, as well as the various transformations that can be performed, focusing on a selection of the most significant and recent advances. By succinctly describing and conveying the versatility of borrowing hydrogen chemistry, we anticipate its uptake will increase across a wider scientific audience, expanding opportunities for further development. (ACS Cent. Sci., 2021, 7, 570-585) [link]
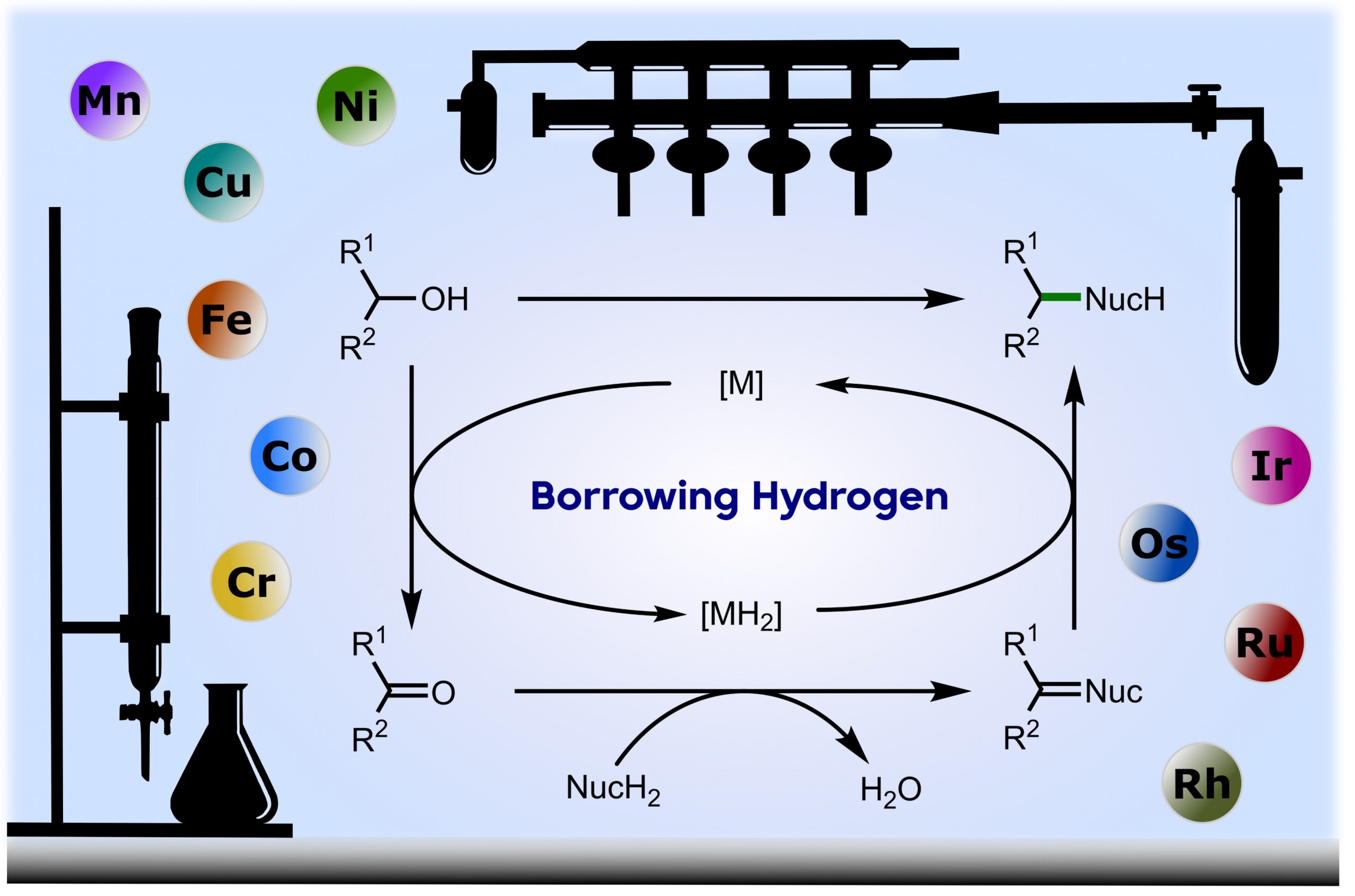
Transition Metal Free α-C-Alkylation of Ketones Using Secondary Alcohols
Alkylation is a fundamental transformation in synthetic chemistry that is routinely performed across the entire spectrum of chemical industries. Traditionally, hazardous alkyl (pseudo)halides are commonly employed for alkylation processes, which can result in non-selective transformations due to multiple alkylation, whilst generating stoichiometric waste products that must be separated from the target compound. As such, the development of selective alkylation methodologies that employ less toxic reagents, whilst generating benign by-products, is an important goal for improving sustainability within the synthetic community.
A base-mediated α-C-alkylation of ketones with secondary alcohols has been developed as an alternative to borrowing hydrogen-type alkylation. This transition metal free approach employs KOt-Bu as the base and exhibits a broad scope, allowing a range of commodity aliphatic secondary alcohols and 1-arylethanols to be employed as alkylating agents. Aryl methyl ketones undergo selective mono-α-C-alkylation in high isolated yields (23 examples, 65% average yield). (Tetrahedron, 2020, 76, 131571) [link] [Invited contribution to the Special Issue on Strategies for Efficient Organic Synthesis Dedicated to the Achievements of Prof. Jonathan Williams]
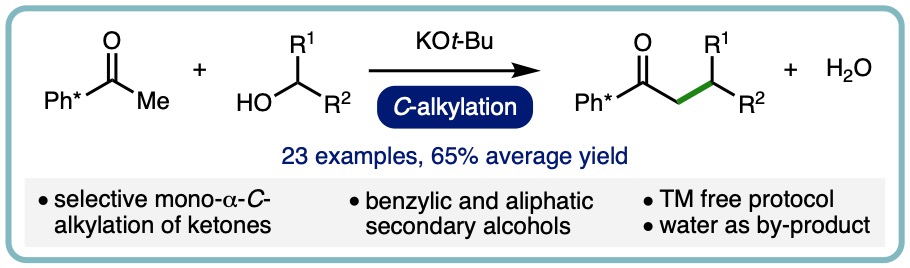
Manganese-Catalyzed One-Pot Conversion of Nitroarenes into N-Methylarylamines Using Methanol
N-methylamines are an important structural motif that feature within a diverse array of useful compounds, including medicines, agrochemicals, dyes and surfactants. More specifically, N-methylarylamines can be found within a broad range of pharmaceuticals, including Rosiglitazone, Clobazam and Osimertinib. Traditional methods for N-methylation of arylamines include the use of hazardous reagents such as methyl iodide, dimethyl sulfate and diazomethane, which typically produce a mixture of mono- and di-methylated arylamines whilst generating stoichiometric quantities of waste. Reductive couplings of arylamines with carbon dioxide or formic acid can also be employed for N-methylation. Alternatively, the borrowing hydrogen approach enables methanol to be employed as the methylating agent for the selective mono-N-methylation of anilines.
A manganese-catalyzed one-pot conversion of nitroarenes into N-methylarylamines has been developed. This transfer hydrogenation method employs a well-defined bench stable Mn PN3P pincer precatalyst in combination with methanol as both the reductant and the C1 source. A selection of commercially available nitroarenes was converted into N-methylarylamines in synthetically useful yields. (Eur. J. Org. Chem., 2020, 1136-1140) [link] [Invited contribution to the YourJOC Talents Special Issue]
Iron-Catalyzed Borrowing Hydrogen β–C(sp3)-Methylation of Alcohols
Allylic alcohols are privileged motifs in synthetic chemistry due to their widespread availability and diverse reactivity profile. An important transformation of allylic alcohols is the redox isomerization to form synthetically useful enolizable carbonyl compounds, which can be performed using metal catalysts or Brønsted base catalysts. The subsequent incorporation of methyl groups via a-C(sp3)-methylation can significantly impact the pharmacological properties of a molecule. Many commonly employed methylation protocols utilize hazardous reagents such as methyl iodide, diazomethane, or dimethyl sulfate. However, recent advances in borrowing hydrogen catalysis have enabled methanol to be employed as an attractive alternative for ketone a-C(sp3)-methylation, using catalysts based on both precious metals and more abundant 3d transition metals.
A one-pot iron-catalyzed conversion of allylic alcohols to a-methyl ketones has been developed. This isomerization-methylation strategy utilized a (cyclopentadienone)iron(0) carbonyl complex as precatalyst and methanol as the C1 source. A diverse range of allylic alcohols undergo isomerization-methylation to form a-methyl ketones in good isolated yields (up to 84% isolated yield) (Org. Lett., 2019, 21, 7914-7918) [link].
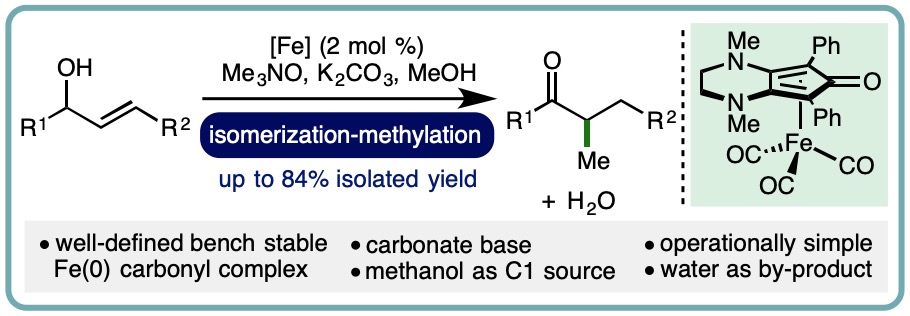
Iron-Catalyzed Borrowing Hydrogen β–C(sp3)-Methylation of Alcohols
The incorporation of methyl groups can have a significant impact upon the pharmacological properties of a molecule. Inspection of Njarđarson’s poster entitled “Top 200 Brand Name Drugs by Prescription in 2016” reveals that a significant proportion contain the C(sp3)−Me motif. As such, the development of new synthetic methods for the direct methylation of C(sp3)−H bonds is an important area of scientific endeavor. Methanol is an attractive reagent for methylation processes. It is an abundant, biodegradable liquid that is less hazardous relative to commonly employed methylation reagents such as diazomethane, dimethyl sulfate and iodomethane.
We report the iron-catalyzed β-C(sp3)-methylation of primary alcohols using methanol as a C1 building block. This borrowing hydrogen approach employs a well-defined bench stable (cyclopentadienone)iron(0) carbonyl complex as precatalyst (5 mol %) and enables a diverse selection of substituted 2-arylethanols to undergo β-C(sp3)-methylation in good isolated yields (24 examples, 65% average yield) (ACS Catal., 2019, 9, 8575-8580) [link] [Highlighted in Synfacts, 2019, 15, 1280] [link]
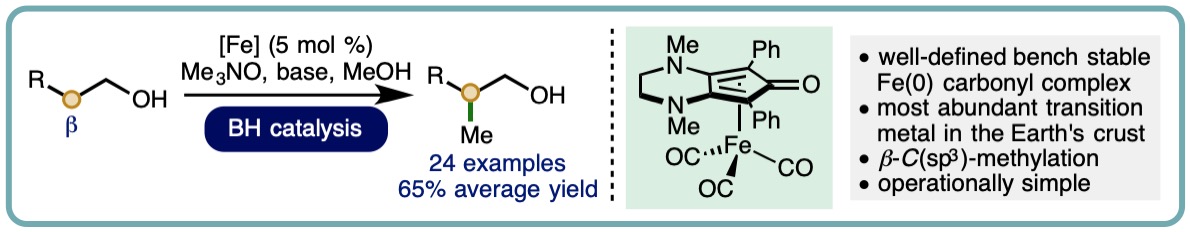
Iron-Catalyzed Borrowing Hydrogen C-Alkylation of Oxindoles Using Alcohols
The oxindole framework is present in a diverse array of naturally occurring compounds. Furthermore, oxindoles that are mono- or disubstituted at the C(3) position are commonly employed in drug discovery programmes, with examples including the development of HIV-1 non-nucleoside reverse transcriptase inhibitors, spircocyclic compounds with anti-cancer and anti-inflammatory properties, and antagonists of progesterone and 5-hydroxytryptamine7 (5-HT7) receptors. The traditional method for alkylation of unprotected oxindoles employs toxic alkyl halides and exhibits poor selectivity (mono- vs. dialkylation, C– vs. N-alkylation) alongside the generation of stoichiometric quantities of undesired byproducts. An alternative approach employs the borrowing hydrogen (BH) principle, also known as hydrogen autotransfer, which allows bench stable and inexpensive alcohols to be used as alkylating agents, generating water as the sole byproduct
A general and efficient iron-catalyzed C-alkylation of oxindoles has been developed. This borrowing hydrogen approach employs a (cyclopentadienone)iron carbonyl complex (2 mol %) and exhibits a broad reaction scope, allowing benzylic and simple primary and secondary aliphatic alcohols to be employed as alkylating agents. A variety of oxindoles undergo selective mono-C(3)-alkylation in good to excellent isolated yields (28 examples, 50-92% yield, 79% average yield) (ChemSusChem, 2019, 12, 2345-2349) [link].
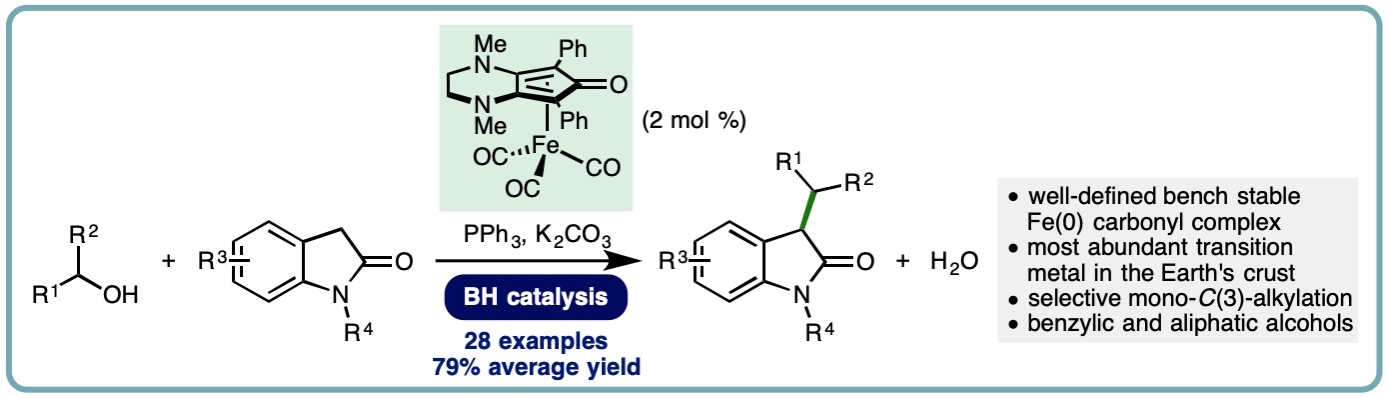
Manganese-Catalyzed N-Alkylation of Sulfonamides Using Alcohols
The sulfonamide functional group is present in a diverse array of bioactive compounds. More specifically, N-alkylsulfonamides are commonly employed in drug discovery programmes. Classical methods for N-alkylsulfonamide synthesis include the reaction of amines with activated sulfonyl derivatives (commonly sulfonyl chlorides), N-alkylation of primary sulfonamides with alkyl halides, and reductive amidation using aldehydes. Drawbacks of these methods include limited availability and stability of specific sulfonyl chlorides, the use of toxic alkylating agents, and the stoichiometric generation of undesired by-products. In contrast, the borrowing hydrogen (BH) approach allows commodity alcohols to be employed as alkylating agents, with water generated as the only by-product. Traditionally, BH processes have employed precious metal catalysts. However, as part of ongoing efforts to reduce our dependence on precious metal catalysts, recent advances have demonstrated the use of earth-abundant first row transition metal catalysts across a variety of borrowing hydrogen processes.
An efficient manganese-catalyzed N-alkylation of sulfonamides has been developed. This borrowing hydrogen approach employs a well-defined and bench stable Mn(I) PNP pincer precatalyst, allowing benzylic and simple primary aliphatic alcohols to be employed as alkylating agents. A diverse range of aryl and alkyl sulfonamides undergoes mono-N-alkylation in excellent isolated yields (32 examples, 85% average yield) (J. Org. Chem., 2019, 84, 3715-3724) [link] [Highlighted in Organic Chemistry Portal] [link].
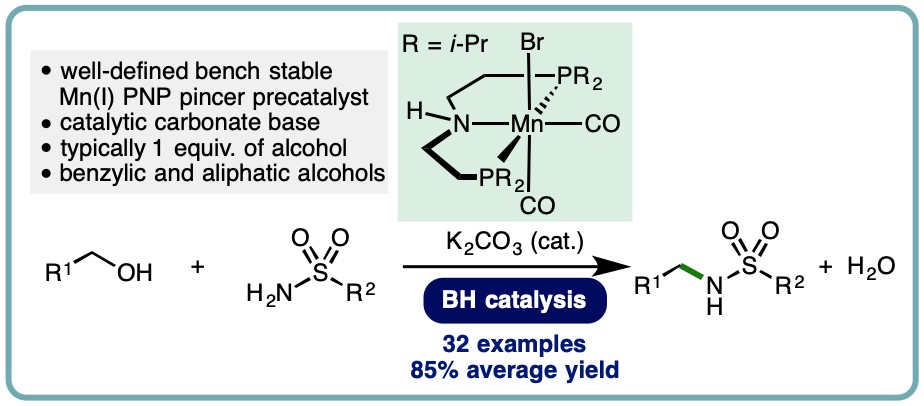
Recent Advances in Homogeneous Borrowing Hydrogen Catalysis Using Earth-Abundant First Row Transition Metals
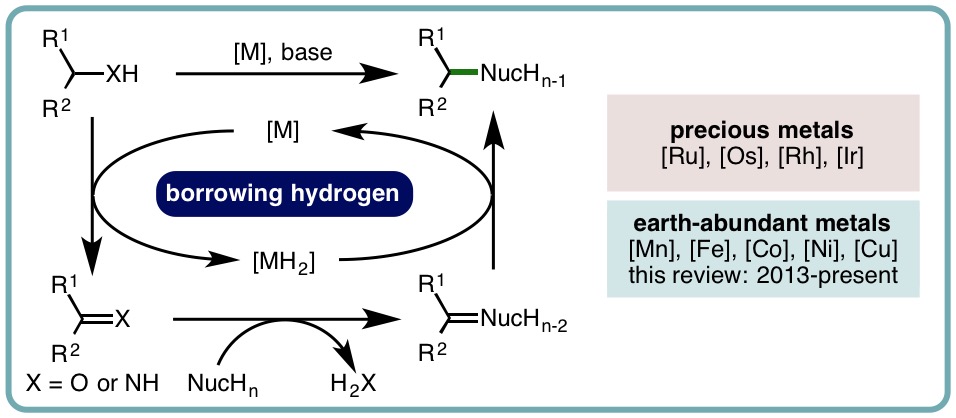
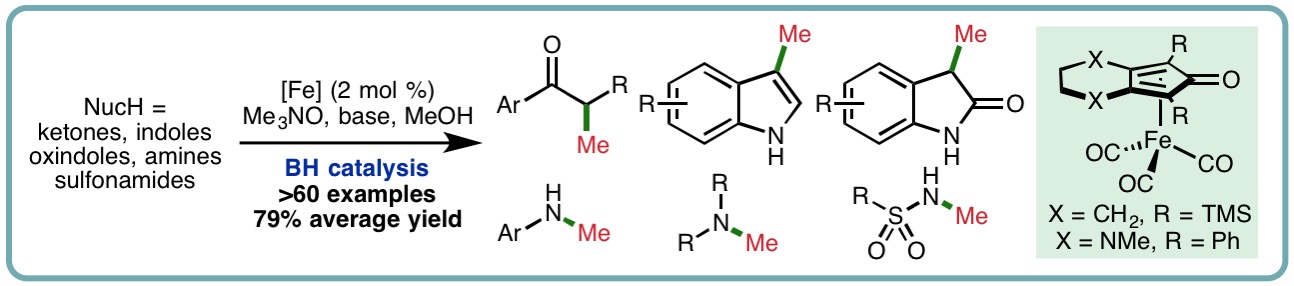
Iron-Catalyzed Methylation using the Borrowing Hydrogen Approach
Methylation is a fundamental transformation in synthetic chemistry that is widely used for the synthesis and functionalization of fine chemicals. Traditional methylation procedures often employ toxic and/or potentially explosive reagents including iodomethane, dimethyl sulfate or diazomethane, among many others. In recent years, methanol, an abundant and biodegradable liquid, has emerged as an attractive alternative reagent for methylation. Borrowing hydrogen (BH), or hydrogen autotransfer, combines a transfer hydrogenation process with a concurrent reaction on the in situ generated reactive intermediate. This one-pot oxidation-reaction-reduction sequence has received much attention due to its inherent high atom economy and minimal waste generation, allowing bench stable and inexpensive alcohols to be used as alkylating agents. In comparison to benzyl and long-chain n-alkyl alcohols, it is challenging to use methanol as the alkylating agent in BH processes, due partly to the increased energy of dehydrogenation (∆H (MeOH) = +84 kJ mol-1, cf. ∆H (EtOH) = +68 kJ mol-1) to form the required transient reactive formaldehyde intermediate.
A general iron-catalyzed methylation has been developed using methanol as a C1 building block. This borrowing hydrogen approach employs a Knölker-type (cyclopentadienone)iron carbonyl complex as catalyst (2 mol %) and exhibits a broad reaction scope. A variety of ketones, indoles, oxindoles, amines and sulfonamides undergo mono- or dimethylation in excellent isolated yields (>60 examples, 79% average yield) (ACS Catal., 2018, 8, 6440-6445) [link].
Exploring Tandem Ruthenium-Catalyzed Hydrogen Transfer and SNAr Chemistry
The carbonyl functional group is one of the most prevalent and versatile in chemistry. However, in some cases, carbonyl compounds suffer from poor stability (e.g., air oxidation of aldehydes to carboxylic acids; enolization leading to deleterious side reactions and erosion of enantiopurity) and limited commercial availability. In comparison, alcohols are typically widely available, inexpensive and relatively inert toward air, moisture and light, making them highly attractive starting materials for synthesis. Furthermore, the alcohol functional group is ubiquitous in pharmaceuticals, agrochemicals, dyes, fragrances, polymers, functional materials and catalysts. As such, the development of novel methods that directly functionalize alcohols, diversifying their reactivity profile, is an important pursuit.
Hydrogen transfer is a powerful approach that can be employed to access the diverse reactivity of carbonyl compounds from alcohol starting materials. Dehydrogenation of secondary alcohol substrates accesses ketones that can react with both nucleophiles and electrophiles (via enolization) in a variety of important reactions including C−C or C−N bond formation. Dehydrogenation of allylic and benzylic alcohols dramatically alters the properties and reactivity of the olefin and arene, respectively, via electronic activation. We have developed a hydrogen transfer strategy for the catalytic functionalization of benzylic alcohols via electronic arene activation, accessing a diverse range of bespoke diaryl ethers and aryl amines in excellent isolated yields (38 examples, 70% average yield). Taking advantage of the hydrogen transfer approach, the oxidation level of the functionalized products can be selected by judicious choice of simple and inexpensive additives (Org. Lett., 2017, 19, 6716-6719) [link].