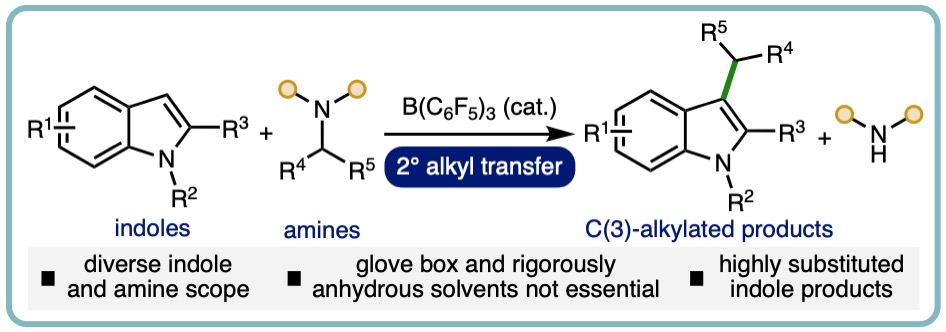
Accessing Highly Substituted Indoles via B(C6F5)3-Catalyzed Sec-ondary Alkyl Group Transfer
Herein, we report a synthetic method to access a range of highly substituted indoles via the B(C6F5)3-catalyzed transfer of 2º alkyl groups from amines. The transition metal-free catalytic approach has been demonstrated across a broad range of indoles and amine 2º alkyl donors, including various substituents on both reacting components, to access useful C(3)-alkylated in-dole products. The alkyl transfer process can be performed using Schlenk line techniques in combination with commercially available B(C6F5)3•nH2O and solvents, which obviates the requirement for specialized equipment (e.g., glove box). (J. Org. Chem., 2024, 89, 4244-4248) [link]
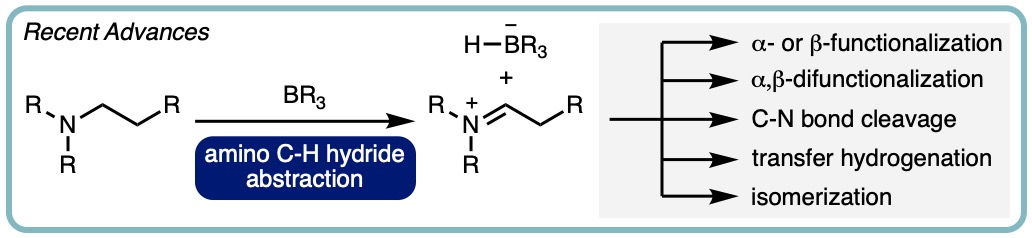
Recent Advances in Catalysis using Organoborane Mediated Hydride Abstraction
C‒H functionalization is widely regarded as an important area in the development of synthetic methodology, enabling the design of more time- and atom-efficient syntheses. The ability of electron-deficient organoboranes to mediate hydride abstraction from α-amino C‒H bonds is therefore of great interest, as the reactive iminium and hydridoborate moieties generated are able to participate in a range of synthetically useful transformations. In this review update, we cover the recent advances made in organoborane mediated hydride abstraction, with particular focus on the catalytic applications of electron deficient boranes for α- or β- functionalization, α,β-difunctionalization, and the dehydrogenation of amines. (Synlett, 2023, 34, 2117-2128) [link] [Invited contribution to celebrate the 60th anniversary of the Matteson homologation] [Highlighted in the 14th EuCheMS Young Investigators Workshop Special Issue]
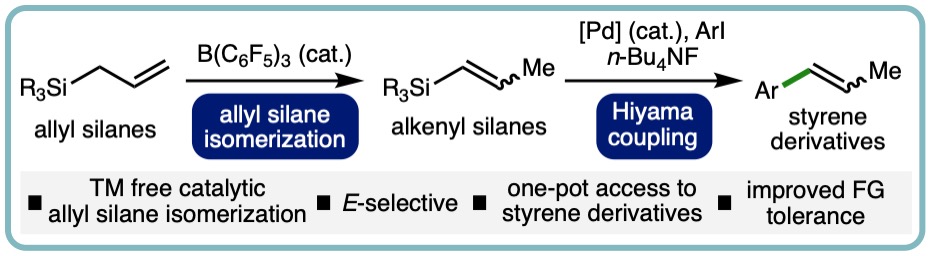
One-Pot Synthesis of Styrene Derivatives from Allyl Silanes via B(C6F5)3-Catalyzed Isomerization – Hiyama Coupling
Alkenyl silanes are useful building blocks in organic synthesis, polymer chemistry, and materials science. They participate in a diverse array of transformations, including electrophilic substitution, polymerization, and cross-coupling reactions. Alkenyl silanes can be accessed by various methods, including nucleophilic substitution of chlorosilanes with alkenyl magnesium reagents, transition metal-catalyzed hydrosilylation of alkynes and allenes, dehydrogenative silylation of alkenes, and Cu-catalyzed silylation of alkenyl iodonium salts. An attractive alternative approach for the formation of substituted alkenyl silanes is via the isomerization of allyl silanes, due to their relative ease of synthesis and commercial availability. A variety of catalytic approaches for the isomerization of allyl silanes to alkenyl silanes have been developed, which employ catalysts based on both precious metals (e.g., Ru, Pd, Ir) and more abundant first row transition metals (e.g., Fe, Co, Ni).
Herein we report a one-pot synthesis of styrene derivatives via a novel B(C6F5)3-catalyzed E-selective isomeriation of readily accessible allyl silanes and subsequent Hiyama coupling of the versatile alkenyl silane intermediates. This one- pot two-step approach enables access to a broad range of styrene derivatives including those containing Lewis basic functional groups that are inaccessible via the previously developed B(C6F5)3-catalyzed isomerization of allyl benzenes. (Org. Lett. 2022, 24, 8694-8697.) [link] [Highlighted in Chemistry Views] [link]
B(C6F5)3-Catalyzed (E)-Selective Isomerization of Alkenes
Alkene-containing compounds are ubiquitous throughout chemistry. Isoeugenol (fragrance), Anethole (food additive), and Licarin A (antimycobacterial) are examples of biologically active molecules that contain internal alkenes. An attractive approach for the formation of internal alkenes is via the isomerization of terminal alkenes, due to their relative ease of synthesis and greater commercial availability. Catalytic approaches to alkene isomerization have been developed that employ a broad range of catalysts based on precious transition metals (e.g., Ru, Rh, Pd, Ir), and more recently earth-abundant 1st row transition metals (e.g., Fe, Co, Ni).
Herein, we report the B(C6F5)3-catalyzed (E)-selective isomerization of alkenes. The transition metal free method is applicable across a diverse array of readily accessible substrates, gaining access to a broad range of synthetically useful products containing versatile stereodefined internal alkenes. The reaction mechanism was investigated using synthetic and computational methods. (Chem. Eur. J., 2022, 28, e202202454). [link]
Electron deficient borane-mediated hydride abstraction in amines: stoichiometric and catalytic processes
The manipulation of amino C–H bonds has garnered significant interest from the synthetic community due to its inherently high atom, step and redox economy. This Tutorial Review summarises the ability of boranes to mediate hydride abstraction from α-amino and γ-amino conjugated C–H bonds. Borane-mediated hydride abstraction results in the generation of reactive iminium hydridoborate salts that participate in a variety of stoichiometric and catalytic processes. The reactions that have utilised this unusual reactivity include those that manipulate amino scaffolds (including dehydrogenation, racemisation, isomerisation, α- and β-functionalisation, and C–N bond cleavage) and those that use amine-based reagents (transfer hydrogenation, and alkylation) (Chem. Soc. Rev., 2021, 50, 3720-3737). [link]

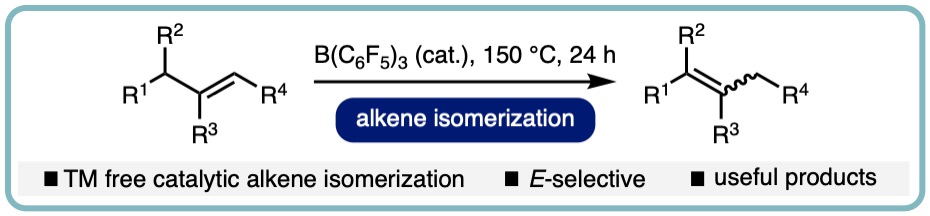
B(C6F5)3-Catalyzed Direct C3 Alkylation of Indoles and Oxindoles
Owing to their intrinsic Lewis acidity, borane catalysts have found numerous applications in synthesis and are traditionally used to activate polarized bonds. Triaryl boranes can also activate unpolarized bonds, such as H–H and Si–H bonds. In a similar vein, we considered if boranes could also be used to cleave heterolytically C(sp3)–H bonds and unveil new approaches to challenging transformations
The direct C3 alkylation of indoles and oxindoles is a challenging transformation and only a few direct methods exist. Utilizing the underexplored ability of triaryl boranes to mediate the heterolytic cleavage of α-nitrogen C–H bonds in amines, in a collaborative research effort with the Pulis and Melen labs, we have developed a catalytic approach for the direct C3 alkylation of a wide range of indoles and oxindoles using amine based alkylating agents. We also employed this borane-catalyzed strategy in an alkylation-ring opening cascade (ACS Catal., 2020, 10, 4835-4840) [link].
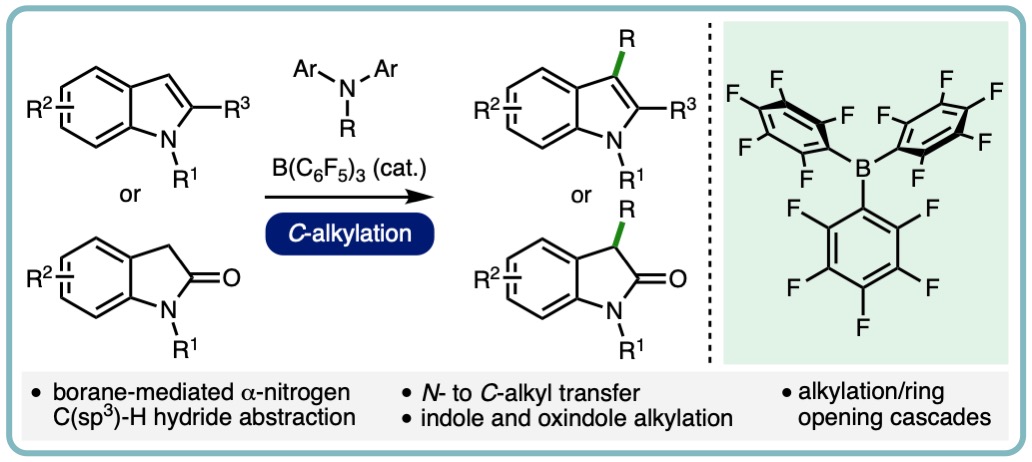
FLP-Catalyzed Transfer Hydrogenation of Silyl Enol Ethers
Silyl enol ethers have often served as a test bed for the development of novel FLP catalytic systems. In contrast to imines and N-heterocycles, which can serve the role of the Lewis base within an FLP-type system, the lower basicity of silyl enol ethers necessitates an additional Lewis base for dihydrogen activation and subsequent hydrogenation.
In a collaborative research effort with the Melen lab, we developed the first catalytic transfer hydrogenation of silyl enol ethers. This metal free approach employs tris(pentafluorophenyl)borane and 2,2,6,6-tetramethylpiperidine (TMP) as a commercially available FLP catalyst system and naturally occurring γ-terpinene as a dihydrogen surrogate. A variety of silyl enol ethers undergo efficient hydrogenation, with the reduced products isolated in excellent yields (29 examples, 82% average yield) (Angew. Chem. Int. Ed., 2018, 57, 12356-12359) [link].
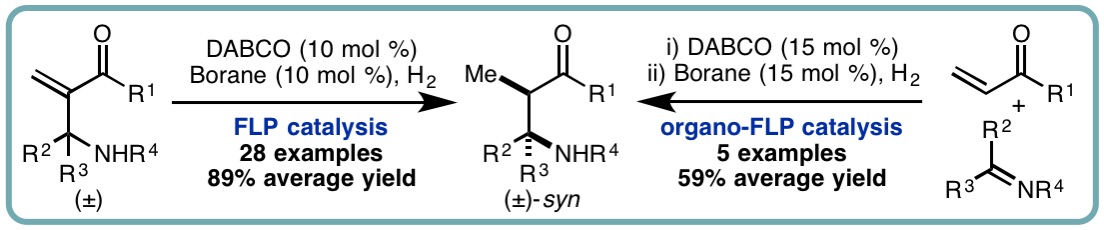
Frustrated Lewis Pair (FLP)-Catalyzed Hydrogenation of Aza-Morita-Baylis-Hillman Adducts and Sequential Organo-FLP Catalysis
Over the past 10 years there has been a surge or research into frustrated Lewis pairs (FLPs). Of particular interest is the ability of FLPs to activate hydrogen for metal-free hydrogenation processes. Despite intensive research efforts, the substrate scope of FLP-catalyzed hydrogenation is somewhat narrow and applications of FLP catalysis in wider organic synthesis remain scarce. FLP-catalyzed hydrogenations of electron-deficient olefins are particularly challenging, often requiring the use of specialized Lewis acidic boranes, out with B(C6F5)3.
In a collaborative research effort with the Melen lab, we developed a metal-free diastereoselective FLP-catalyzed hydrogenation of aza-Morita-Baylis-Hillman (aza-MBH) adducts, accessing a diverse range of stereodefined β-amino acid derivatives in excellent isolated yields (28 examples, 89% average yield, up to 90:10 d.r.). Furthermore, the first example of sequential organo-FLP catalysis has been developed. An initial organocatalyzed aza-MBH reaction followed by in situ FLP formation and hydrogenation of the electron deficient α,β-unsaturated carbonyl compounds can be performed in one-pot, using DABCO as the Lewis base in both catalytic steps (ACS Catal., 2017, 7, 7748-7752) [link].